同分异构现象及其分类
构造异构 碳干异构
(原子间连接的方式和顺序) 位置异构
官能团异构-醇和醚、醛和酮
互变异构-酮式和烯醇式
同分异构 构型异构 顺反异构-烯烃(SP2)
(空间的排布方式) 对映异构-手性分子(SP3)
构象异构 碳链的构象
(C-C自由转动) 环的构象
1、构造异构:
键合原子之间的连接方式和顺序。按照异构一之间的关系可以分为:
(1)碳干异构:碳链的形状不同。烷烃的异构现象主要是碳干异构,也是其它各类化合物构造异构的基础。如:戊烷有三个异构体,丁醛有两个异构体
(2)位置异构:碳链形状不变,而官能团或者取代基在碳链上的位置不同。如:二甲苯由3种异构体,正戊烷有3种一氯代产物。
(3)官能团异构:分子式相同,官能团不同。如:醇与醚、醛与酮、酸与酯互为官能团异构体。
(4)互变异构:特殊的官能团异构现象,异构体之间可以迅速的相互转化,共存互变、不可分离是互变异构体系的特点。最重要的互变异构现象是:

如:

再如:酚虽然属于烯醇式,但也可以通过酮式进行反应—Koble反应:
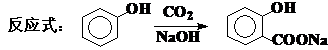
(5) 分子的不饱和度W与结构特征(构造)
对于分子式:IVnImIIxIIIy (IV为四价元素、I为一价元素、II为二价元素、III为三价元素,m、n、x、y分别表示相应的原子数目),
W = (2n+2-m+y) ¸2 二价元素与W无关!
不饱和度W所代表的结构特征:
W=1:一个双键(C=C、C=O、C=N、N=O…等) 或一个环(三、四、五、六元环…等,既可以是碳环,也可以是杂环)
W=2:一个三键(CºC或CºN) 或2个双键,或者2个环,或者1个双键+1个环。
W³4:首先考虑分子含苯环,苯环的W=4 !
练习:写出分子式C4H8O代表的所有化合物,并正确命名。
3、共振论的应用:
分子或活性中间体内能
分子或活性中间体的内能在很大程度上决定了反应的活性、方向和产物的分配,在研究和讨论反应时,经常遇到此类问题。使用共振论定性解释这类问题时,规则简单,结论明确。定性结论主要有两条:
①具有较多共振极限式的体系内能较低;
②包含一个内能特别低的共振极限式的体系内能较低,反之包含一个内能特别高的共
振极限式的体系内能较高。
例如:氯苯硝化反应活性和定位规则的解释
硝化是亲电取代反应,混酸中的有效亲电试剂是硝基正离子+NO2。当亲电试剂+NO2进攻苯环时,有效取向可以是Cl的邻位、间位和对位,将生成三种正离子中间体,应用共振论可以比较这三个中间体的内能,中间体内能较低的反应活性高(能垒小、速度快),将生成主要产物。
应用结论①:当发生邻位取代时,中间体的共振极限式可以写出四个,而间位取代时,中间体的共振极限式只有三个,所以前者内能较低(较稳定),反应速度快,相应的产物为主要产物。
应用结论②:邻位取代时,中间体的共振极限式中含有一个内能特别低的共振极限式(a),正电荷携带在Cl原子上,在a式中,除H之外,所有的原子(包括Cl)都满足八电子,因此这个极限式的内能特别低,所以邻位取代形成的中间体内能较低,相应的产物是主要产物。
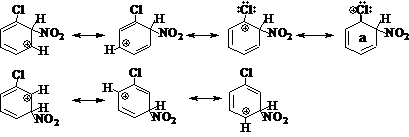
对位取代的情况与邻位完全相同(自己完成),所以Cl为邻、对位定位基。
共振与互变异构的区别:
判断标准:是否发生了原子的迁移!
判断下列各式表示的是共振?还是互变异构?
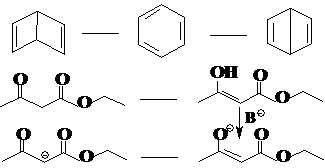
2、共振极限式内能高低的比较
(1)等价的共振极限式对真实分子的贡献相等,不等价的极限式对真实分子的贡献不同,内能较低的极限式对真实分子的贡献较大。如:苯分子的5共振极限式中,1和2等
价,内能较低,对真实分子的贡献各为39%,而3、4和5等价,内能较高,对真实分子的贡献各为7.33%。
(2)真实分子的内能低于每一个共振极限式,因此共振是一种稳定化作用。如:真实的苯分子的内能比它的5个共振极限式中的每一个所代表的内能都低。共振论把真实分子的内能与内能最低的共振极限式之间的能量差别定义为该分子的共振能。苯分子的共振能为:150.4kj/mol
(3)电荷分离的共振极限式(称为极性极限式)内能较高,对体系的贡献较小;多于2个表观电荷的极性极限式内能更高,对真实分子的贡献更小,在相邻原子上出现相同电荷的极性极限式非常高,对中性分子的贡献可以完全忽略不计。如:
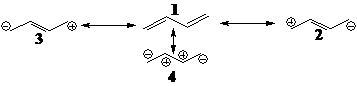
其中4式对丁二烯的贡献可以忽略,而2式和3式的贡献也比1式小得多,真实的丁二烯分子主要与1式相似。
(4)情况相同时,负电荷携带在电负性较大的原子上的极限式内能较低,而正电荷携带在电负性较小的原子上的极限式内能较低。如:
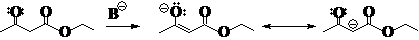
注意:乙酰乙酸乙酯(EAA)在碱性条件下形成的负离子,负电荷在氧原子上更合理(因为氧原子的电负性比碳大得多,更容易携带负电荷),极限式2比1的内能低,因此2的贡献比1大。所以在书写反应机理时,遇到这种离子,写成2更合理。
但这个规则不能与经典价键理论的八电子偶规则相抵触,例如:
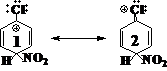
尽管Cl的电负性比C大,但极限式2比1的内能低,这是因为后者除H之外所有的原子都满足八电子,而前者含有一个6电子的C+。
(5)键角或键长偏离正常值的共振极限式内能较高,对真实分子的贡献很小。如:对于苯分子,杜瓦极限式的键长和键角都大大偏离了正常范围,因此它们的内能比凯库勒极限式高得多,对真实苯分子的贡献只有7.33%,而凯库勒极限式分别为39%。
含有离域键的分子用一个经典结构式不能准确表示,如:苯分子的凯库勒结构式(实际是环己三烯)并不能准确地表示真实的苯分子,因此它只是苯分子的一个代表符号。共振论的基础与分子轨道理论相同(也是量子力学和数学方法),区别在于:分子轨道理论使用原子轨道组合成分子轨道,而共振论采用经典结构式的组合来表示分子。
共振论的提出者-鲍林(L.Pauling)认为:苯分子是以下五个结构的组合:
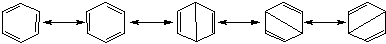
y1 y2 y3 y4 y5
y = c1y1+c2y2+c3y3+c4y4+c5y5 其中:C1=C2=0.39,C3=C4=C5=0.0733
在共振论中,经典结构式称为共振极限式,y1和y2 为凯库勒极限式,y3、y4和y5为杜瓦极限式。
1、共振极限式的书写规则:
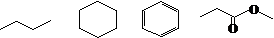
(1)所有的共振极限式(canonical forms)都必须是严格的经典路易斯结构式(Lewis structures),也就是必须符合经典价键理论。目前有机化学中使用的结构简式都是路易斯结构式,如:
(2)在同一分子的所有共振极限式中,原子核都不能发生移动,能够移动或重新排布的是离域电子(一般指π电子或孤对电子,在讨论超共轭效应时也可以是C-H键的σ电子),如氯苯分子可以表示为:
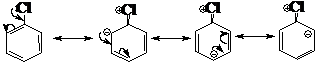
这可以说明在氯苯的环上,邻位和对位较负(电子云密度较高),所以亲电取代反应发生在邻对位。
(3)共振涉及的所有原子必须共平面或接近共平面。有时,原子和原子团之间的立体排斥作用可能破坏分子的平面性,共振作用就会变弱,或者完全不存在。如:
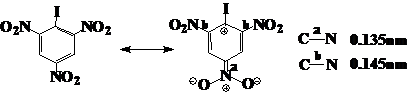
这说明:对位硝基参入共振,而邻位硝基并不参入共振。
(4)在同一个分子的各个共振极限式中,单电子(孤电子)数和电子对数都不能改变。注意:在Lewis结构式中,一个表观负电荷相当于一对电子。如:

5、前线轨道理论(FMO)─(福井谦):
化学反应的本质是电子转移。最容易的电子转移必然是:发生在:电子从最高占据轨道转移到最低空轨道。所以,从电子转移的角度说,化学反应只有一种可能的反应模式即:
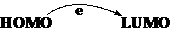
前线轨道理论的应用-环加成反应:
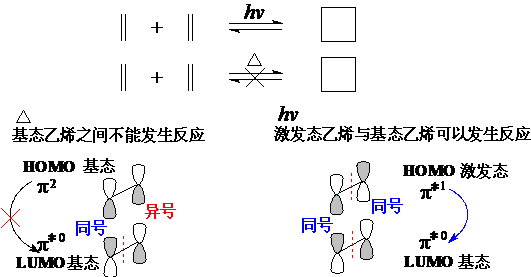
反应条件:加热△与光照hn虽然都是提供能量的手段,但它们绝然不同。
热量是连续的物理量、分子中的电子能是量子化的物理量,按照物理学的基本原理,这两类不同性质的物理量不同相互转化,因此在加热条件下,电子不可能被激发,分子处于基态。
而光能是量子化的物理量,在光照条件下,电子可以吸收相应的光能被激发,体系中既有基态分子,也有激发态分子(但体系中的分子不可能同时被激发)。
激发态分子的HOMO 和 LUMO与基态分子不同,如:
分子的 HOMO 和 LUMO与反应的条件相关:
乙烯 丁二烯 苯
HOMO 基态(△): p ψ2 ψ2 或 ψ3
激发态(hn): p* ψ3* ψ4*或ψ5*
LUMO 基态(△): p* ψ3* ψ4*或ψ5*
激发态(hn): σ* ψ4* ψ5*或ψ4*
在环加成反应中,如果一个分子的HOMO和另一个分子的LUMO的节面都是奇数(或偶数),那么它们可以实现同号重迭,两个分子的MO之间不存在节面,电子可以顺利发生转移,反应则是允许的,否则是禁阻的。
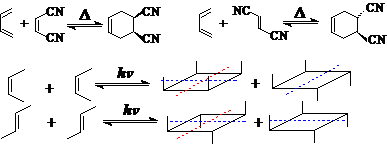
如: 4 + 2 反应(Alder-Diels反应,加热允许,hv禁阻):
在加热条件下,丁二烯和乙烯体系都处于基态,丁二烯的HOMO是y2 ,只有1个节面,乙烯的LUMO是p*,也只有1个节面,二者节面都是奇数,所以反应是加热允许的。当然,也可以用丁二烯的LUMO-y*3和乙烯的HOMO-p来判断,二者都有2个节面,节面同为偶数,也是加热允许的:
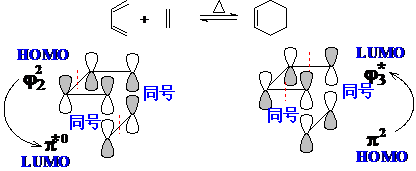
在光照条件下,如果丁二烯被激发,其HOMO为y3*1,而乙烯处于基态,其LUMO为p*,前者有2个节面,后者只有1个节面,所以是禁阻的;如果乙烯被激发,只能使用其HOMO-p*1,而基态丁二烯必然使用其LUMO-y3*0,同样是禁阻的:
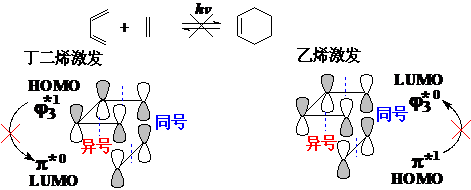
2+2反应(hv允许,加热禁阻)
在加热条件下,乙烯都处于基态,一个分子的HOMO(为p)与另一个分子的LUMO(为p*),节面数目偶奇不同,所以2+2反应加热是禁阻的。
在光照条件下,体系中既有基态乙烯分子(尚未吸收光能),也有激发态乙烯分子(已经吸收光能),反应可以在激发态乙烯的HOMO-p*1与基态乙烯分子的LUMO-p*0之间进行,二者相同,当然节面都是奇数(1个),所以2+2反应是光照允许的。
环加成反应的选择性:
①烯烃构型保持:4+2和2+2都是同面-同面反应,平面内的构型在反应中保留。
②4+2反应次级效应和空间效应:endo和exo
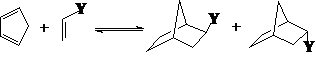
exo endo
Y饱和 主 次 (空间效应)
Y不饱和 次 主 (次级效应)
如:
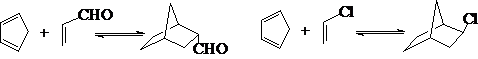
③4+2的区域选择性──邻对位规则

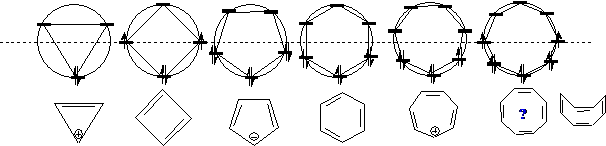
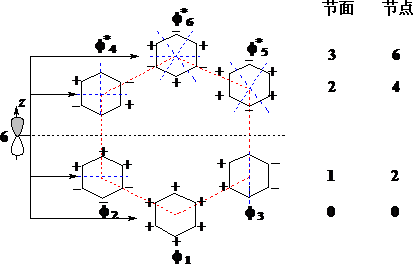
4、休克尔芳香规则:
平面轮烯的芳香性一直是化学家感兴趣的问题。
平面轮烯的p电子总数:= 4n + 2 芳香性 -特别稳定
= 4n 反芳香性-特别不稳定
= 其它 非芳香性-与普通体系相似
环状闭合共轭体系的休克尔分子轨道处于圆的内接正多边形的各个顶点,圆心的能量水平为原子轨道PZ的能量。如环丁二烯、苯等。
如:环戊二烯体系:
3、休克尔(HÜckel)分子轨道-HMO:
在LCAO法中,休克尔分子轨道专门处理分子的p键体系,是最近似、使用最广泛的分子轨道。
原则:s键构成的分子骨架用杂化轨道理论处理,p键体系(即剩余的p轨道)用LCAO分子轨道法处理。
乙烯:两个C原子都是SP2,4个C-H s键和1个C-C s键构成了乙烯的分子平面
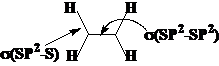
每一个C都剩余一个垂直于分子平面的PZ轨道,由这两个PZ轨道组合成乙烯的p-分子轨道,即休克尔分子轨道-HMO:

每个PZ轨道上有1个电子,共有2个P电子,都填充在p上,所以乙烯的p-电子构型为p2p*0。这两个p-电子与原来的2个P-电子相比,能量下降,所以形成稳定分子。
丁二烯:4个C都是SP2,除了s骨架之外,每个C都有一个PZ轨道和1个P-电子
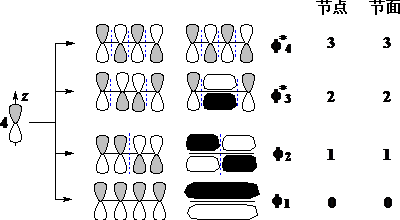
丁二烯的p-电子构型为:f12 f2 2f3*0 f4*0
苯:6个C都是SP2杂化,s骨架构成平面正六边形,完全符合杂化轨道理论。此外,
每个C都有一个PZ轨道和1个P-电子,由它们组合成苯的HMO。
苯分子的p电子构型为:f12f22f32f*40f*50f*60
2、组合结果:数目相等、能量分裂
①数目相等:分子轨道和参与组合的原子轨道数目相等,即:n 个原子轨道必定组合成 n 个分子轨道。
②能量分裂:原子轨道组合成分子轨道以后,能量发生分裂。有的分子轨道能量降低-成键分子轨道,有的能量下降-反键分子轨道(以*标出),有的能量不变-非键分子轨道。
在LCAO法中,能量分裂表现为对称分裂,从而维持体系总能量守恒。
分子轨道的能量高低与节面(电子云密度等于零的平面)或节点(节面与键轴的交点)的数目相关:节面越多,能量越高,同一个分子的分子轨道,节面按自然数变化(0、1、2、3…)
分子轨道理论完全抛开了经典价键理论,以量子力学为基础、数学为手段来认识分子的结构,不存在价键、化合价、公用电子对等概念。简单地说:分子轨道理论把分子当成原子来处理。
-5-
分子轨道理论认为:原子在组成分子时,首先由原子轨道组合成分子轨道,然后原子体系的电子全部填充在分子轨道中,如果能量下降,则形成稳定的中性分子,否则不能形成分子。
这个概念的关键词是:组合和填充
填充:电子在分子轨道体系的填充规律与原子核外电子排布完全相同,遵循相同的规律和规则:①能量最低原理;②保利不相容原理(1个轨道最多填充2个电子);③洪特规则(半满规则)。
组合:既可以根据薛定锷方程,讨论分子的完整准确的分子轨道(可惜只适用于少数简单分子),也可以引于近似条件,讨论分子轨道。由于各种近似条件和近似方法不同,所以出现了各种不同形式的分子轨道。其中最简单、最常用的组合方式是:原子轨道线性组合法-LCAO。
以下我们主要讨论LCAO法
1、组合条件:
①能量接近:
组合使用的原子轨道之间能量越接近,组合越有效(形成的分子内能较低),如果能量差别太大,可能变成无效组合(形成的分子内能叫高或者不稳定)。如:能量相等的2Pz轨道组合成的C=C和C=O中的π键,内能较低,比较稳定,而有能量差别很大的2Pz和3Pz组合成的C=S内能较高,所以硫醛、硫酮类化合物绝大部分都不稳定,很难制备。
②对称性一致:
两个Pz轨道对Z轴都是轴对称的(绕Z轴一周,±符号相同,记为S),对X轴都是反对称的(绕X轴一周,±符号相反,记为AS)。我们说两个Pz轨道的对称性一致,可以组合成分子轨道。
而1个Pz轨道和1个PX轨道的情况则不同。Pz轨道对于Z轴为S,对于X轴为AS,而PX轨道相反,它对于Z轴为AS,对于X轴为S,我们则认为二者的对称性不一致,不能组合成分子轨道。
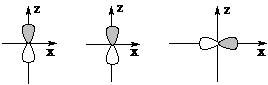
对于Z轴 S S AS
对于X轴 AS AS S
Pz 与Pz 对称性一致,可以组合
Pz与 PX 对称性不同,不能组合
对称性一致体现了共价键的方向性,可以解释分子的立体形状。
3、杂化轨道的应用:
共价键的饱和性和方向性
分子的立体形状:分子的立体形状取决于观察中心C的杂化方式
例如:3-戊烯-1-炔
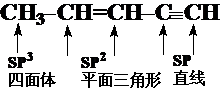
丙二烯的中心C参入形成两个p键,是SP杂化。
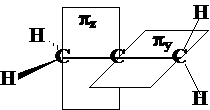
共价键的轨道构成,如:3-戊烯-1-炔
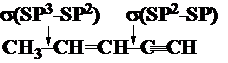
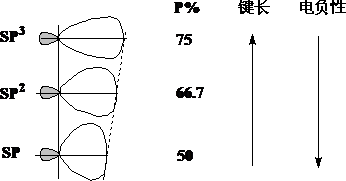
杂化方式与s单键键长:P成分越多,单键越长。
杂化方式与碳原子的电负性:P成分越多,电负性越小;S成分越多,电负性越大
湖北省互联网违法和不良信息举报平台 | 网上有害信息举报专区 | 电信诈骗举报专区 | 涉历史虚无主义有害信息举报专区 | 涉企侵权举报专区
违法和不良信息举报电话:027-86699610 举报邮箱:58377363@163.com